Pulmonary Hypertension
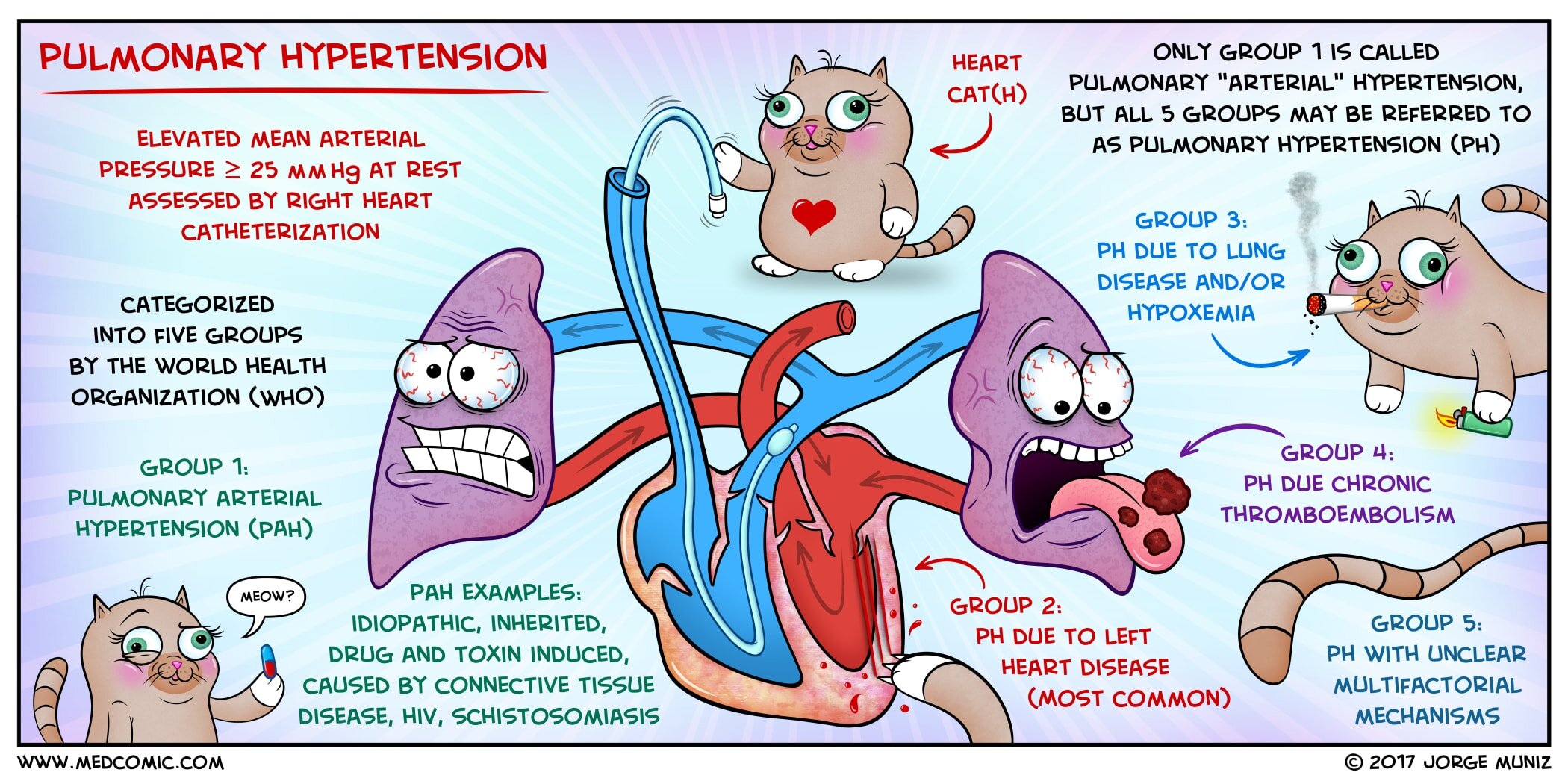
Pulmonary hypertension (PH) refers to increased pressure in the pulmonary circulation. It develops when pulmonary vessels become constricted and/or obstructed, which can occur in a wide variety of conditions. The increase in pressure is measured by right catheterization, and is defined as a mean pulmonary arterial pressure ≥ 25 mm Hg at rest.
PH leads to right ventricular hypertrophy and enlargement as the ventricle pumps against increased resistance. Right-sided heart failure (cor pulmonale) can eventually develop. Signs and symptoms of PH include dyspnea on exertion, fatigue, substernal chest pain, syncope, hepatomegaly, neck vein distention, and leg edema. Narrow splitting of S2 with an accentuated pulmonic component (P2) may be heard on auscultation.
Echocardiography is a helpful initial test to estimate the mean arterial pressure and may also demonstrate enlargement of the right ventricle and atrium, with tricuspid regurgitation. Echocardiography also allows for an assessment of left ventricular function, valvular disease, and congenital heart disease. The mean arterial pressure is definitively diagnosed with right heart catheterization.
The World Health Organization (WHO) categorizes PH into 5 groups and the management of PH depends on the etiology. Possible underlying causes should be identified and treated. Treatment options include oxygen therapy, chronic anticoagulation, calcium channel blockers, prostacyclin (vasodilator), PDE5 inhibitors, and heart-lung transplantation in severe cases.